


What is Amyotrophic Lateral Sclerosis (ALS)?
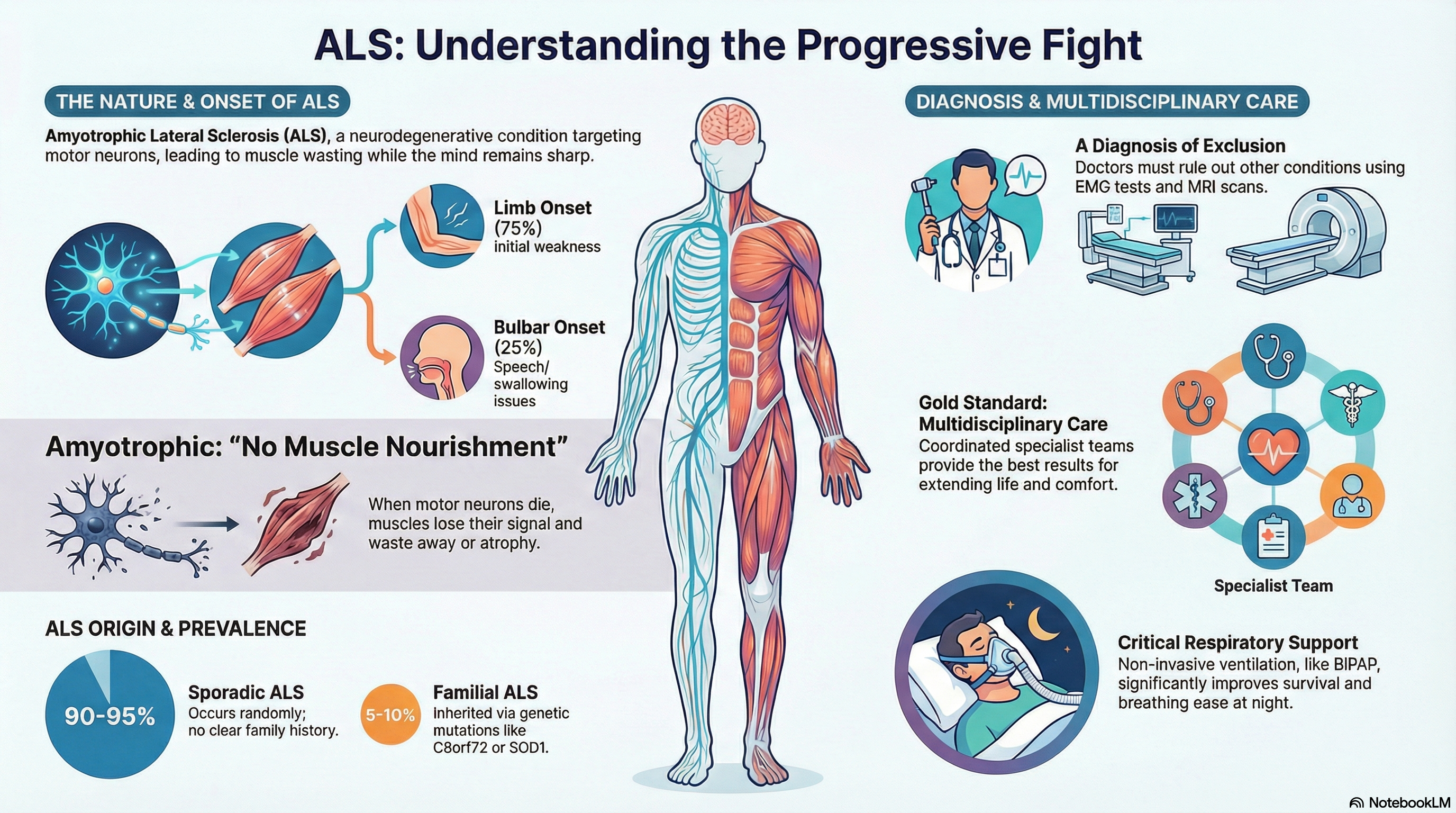
Amyotrophic Lateral Sclerosis (ALS), frequently referred to as Lou Gehrig’s Disease, is a progressive and terminal neurodegenerative disease. It specifically targets the motor neurons—nerve cells in the brain and spinal cord that control voluntary muscle movements like walking, talking, and breathing.
The name describes the disease's impact on the body:
-
Amyotrophic: "No muscle nourishment." When motor neurons die, they stop sending signals to the muscles, causing them to waste away (atrophy).
-
Lateral: Refers to the specific areas in the spinal cord where these nerve cells are located.
-
Sclerosis: As these areas degenerate, they lead to "hardening" or scarring.
In ALS, while the body loses the ability to initiate and control muscle movement, the senses (sight, smell, hearing, taste, and touch) usually remain intact, and for many, the mind remains sharp.
Causes of ALS
The exact cause of ALS is not yet fully understood, though researchers believe it is a combination of genetic and environmental factors.
Genetic Categories
-
Sporadic ALS (90-95% of cases): The most common form occurs randomly without a clear family history. Its cause remains unknown.
-
Familial ALS (5-10% of cases): This form is inherited. The most common genetic link is a mutation in the C9orf72 gene, followed by SOD1, TARDBP, and FUS.
Risk Factors and Triggers
-
Age and Sex: Onset typically occurs between ages 40 and 70. In younger populations, men are slightly more likely to be diagnosed, though this gap narrows with age.
-
Military Service: U.S. veterans are diagnosed at nearly twice the rate of the general population, though the reason is still under investigation.
-
Cellular Dysfunction: In ALS patients, the "internal machinery" of the nerve cells fails. This includes the buildup of toxic proteins, abnormalities in RNA metabolism, and the breakdown of the cell's transport system (cytoskeleton).
Symptoms of ALS
The onset of ALS is often subtle and varies depending on which motor neurons are affected first.
Common Early Signs
-
Limb Onset: Approximately 75% of people first notice weakness in their arms or legs. This can manifest as "split hand syndrome," tripping, or dropping objects. You may also notice fasciculations (painless muscle twitches).
-
Bulbar Onset: About 25% of cases begin with speech or swallowing difficulties, such as slurred speech or a nasal-sounding voice.
Disease Progression As the disease spreads, symptoms become more widespread:
-
Total Muscle Loss: Eventually affecting all limbs and the trunk, leading to an inability to walk or stand.
-
Respiratory Failure: The weakening of the diaphragm and chest muscles makes breathing difficult. This is the most serious complication of ALS.
-
Cognitive Changes: While most maintain sharp thinking, up to 60% experience mild behavioral changes, and 15% develop Frontotemporal Dementia (FTD).
-
Pseudobulbar Affect: Involuntary, uncontrollable episodes of laughing or crying.
Diagnosis of ALS
Diagnosis is challenging because no single test can confirm ALS. It is often a "diagnosis of exclusion," where other conditions like herniated disks or multiple sclerosis must be ruled out first.
The Diagnostic Workup
-
Neurological Exam: A doctor looks for "Upper Motor Neuron" signs (stiffness and brisk reflexes) and "Lower Motor Neuron" signs (muscle wasting and twitching).
-
Electromyography (EMG): This is a critical test that measures the electrical activity of your muscles to confirm nerve damage.
-
MRI Scans: Used to ensure that structural issues in the brain or spinal cord are not causing the symptoms.
-
Clinical Criteria: Doctors traditionally use the El Escorial or the simplified Gold Coast criteria to determine if motor neuron loss is present across different body regions (such as the throat, arms, and legs).
Treatment of ALS
While ALS is currently incurable, multidisciplinary care—coordinated by a team of specialists—is the gold standard for extending life and improving comfort.
Disease-Modifying Medications
-
Riluzole: An oral medication that can modestly extend survival by a few months.
-
Edaravone: An antioxidant treatment (IV or oral) that may slow the decline of physical function in certain patients.
-
Sodium Phenylbutyrate/Taurursodiol: A combination therapy approved to slow disease progression and prolong life.
-
Tofersen: A targeted gene therapy specifically for patients with the SOD1 mutation.
Supportive and Palliative Care
-
Respiratory Support: Non-invasive ventilation (NIV), such as a BiPAP machine, helps patients breathe more easily at night and significantly improves survival.
-
Nutritional Support: If swallowing becomes dangerous, a gastrostomy (PEG) tube can ensure adequate nutrition and hydration.
-
Therapies: Physical, occupational, and speech therapies provide equipment (like eye-gaze communication devices) and strategies to maintain independence for as long as possible.
Prevention of ALS
Because the majority of ALS cases occur sporadically and have a strong genetic component, there is currently no proven way to prevent the disease.
Risk Reduction Research While not definitive, research into environmental factors suggests that avoiding high-dose exposure to certain toxins, such as lead or specific pesticides, may be beneficial for general neurological health.
Genetic Counseling For families with a known history of ALS, genetic counseling and testing can help individuals understand their risk and, in some cases, provide access to early-intervention clinical trials or targeted gene therapies like Tofersen before symptoms become severe.
Multidisciplinary Monitoring For those already diagnosed, the focus shifts to "secondary prevention"—preventing complications. This includes early placement of feeding tubes to prevent malnutrition and early use of breathing support to prevent respiratory crisis.


